The HEARTFELT Study is a pragmatic, single-blind, randomized crossover trial testing the effectiveness of autonomous remote patient peripheral edema monitoring and reporting in HEART FailurE compared with conventionaL remoTe patient monitoring.
Primary objective
Establish whether the Heartfelt device is safe to use and effective at reducing heart failure hospitalizations (HFHs).
Study size and follow-up
- 1,500 patients, plus 100 “Vanguard” patients for operational setup
- Main trial of 2 x 6 months, with randomized crossover between intervention and control at 6 months
- Long-term follow-up of up to 4 years after the main trial
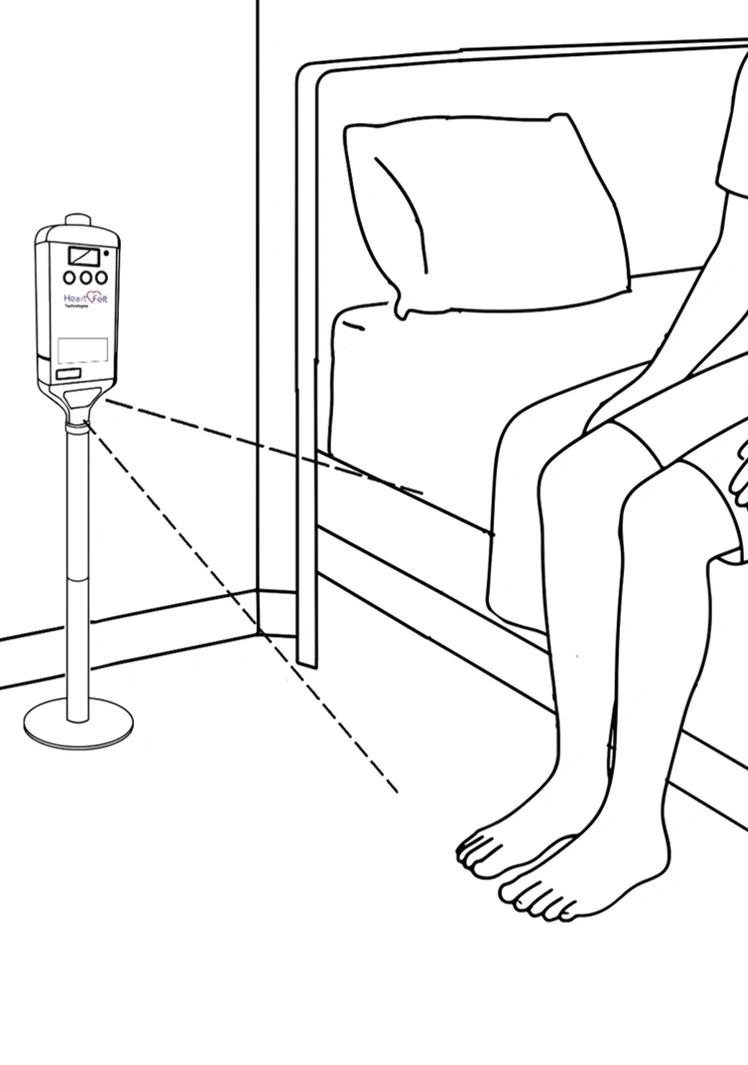
What is the Heartfelt Device?
The Heartfelt Device is a small, non-invasive home-based sensor that measures the volume of a patient’s feet and lower legs as they walk past it.
- There is no need to touch the device, stand on it, press any buttons, or wear anything.
- It works in the background while the patient gets on with their day.
- The device securely sends measurements to the study team.
If the device detects that a patient’s peripheral edema is increasing, it can send a health alert to the clinical team. That may lead to a phone call or review to check symptoms and, if needed, adjust treatment.
Because it works automatically, it can collect much more health information than normal home monitoring.
The Heartfelt Device
Patient Detection
The Heartfelt Device scans a three-metre radius.
Patient Tracking with AI
AI technology tracks a patient as they move around the room to capture 360-degree imagery.
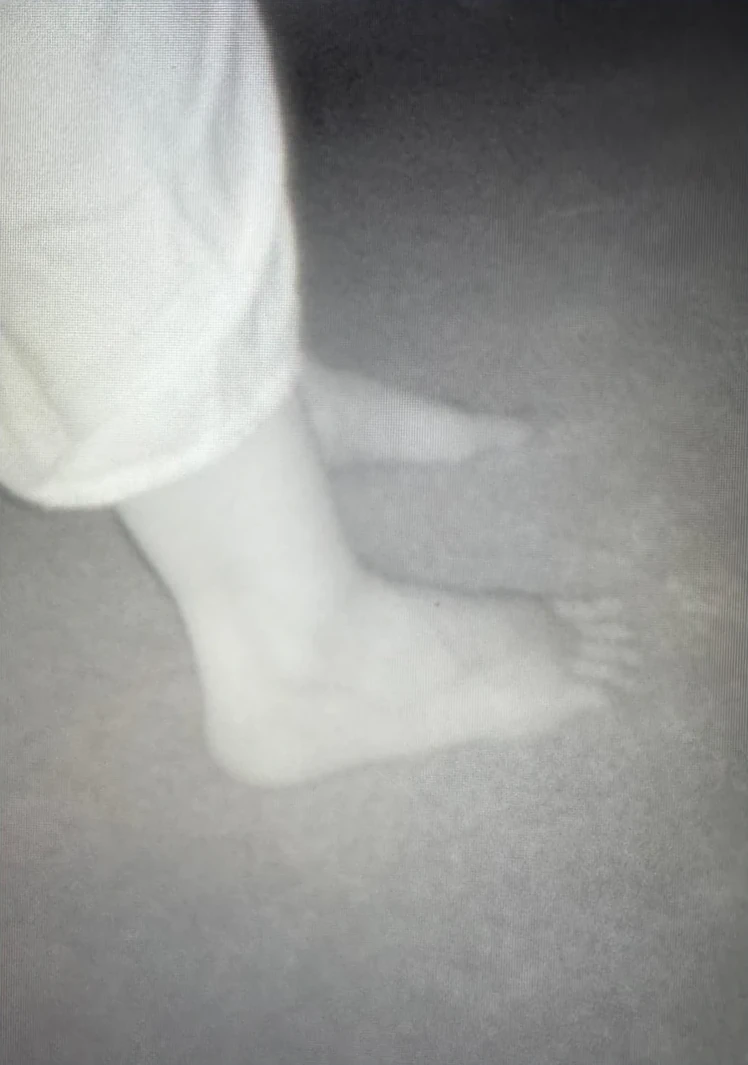
Image Capture
The scanner takes 1,200 pictures a minute to create a 3D image.
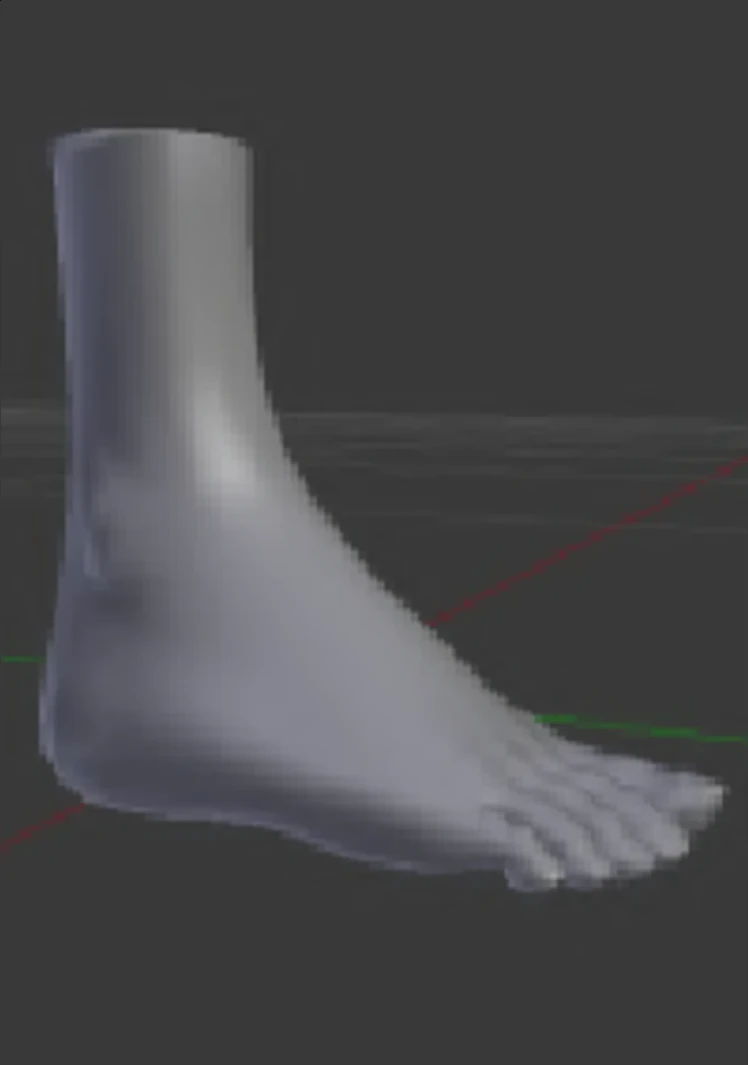
Foot Volume Calculation
A virtual "tank" of water around the 3D foot assesses its volume.

Alert and Action
If the foot volume gets too high, indicating fluid retention, an alert is sent and action is taken if the patient is on the interventional arm.
The data behind the device
47% reduction in HF events
Average 13 days advance warning of hospitalization
85-89% sensitivity
92% of patients would recommend the Heartfelt device to a friend
A solution for the patients you have, not the patients you want
Study description
This study looks at the effect of using a remote patient monitoring device, the Heartfelt device, with health alerts to monitor the development of peripheral edema in patients with heart failure (HF). The hypothesis is that this passive measurement method will lead to better data availability, which in turn will improve patient care and reduce hospitalizations for the management of worsening HF (HFHs).
Participants will be recruited through Remote Patient Monitoring Providers (RPMPs) in the United States, using crossover randomization between standard care and Heartfelt device usage with health alerts sent to RPMPs. The RPMPs will follow a protocolized intervention when responding to raised alerts.
Study flow
The schematic below summarizes the screening, enrollment, randomization, and follow-up structure for the study.
Scrollable flowchart showing the same content summarised above.
Flowchart summary
- Start with patients who have had at least two heart failure events over the past 24 months, with one hospitalization that reported peripheral edema, and at least one event in the past 6 months.
- Filter using historical HF and all-cause hospitalizations reported by RPM adherence over the 180 days before screening. Use adherence thresholds of [0-25%), [25-50%), [50-75%), and [75-100%). Adjudicate a maximum of 300 events per bin and, if a bin contains more than 300 events, randomly sample events in that bin to establish the percentage that are HF-related.
- Split patients by adherence bin. If adherence is
>=50%, exclude and report HF and all-cause hospitalizations over 12 months from screening for that bin. If adherence is<50%, apply the inclusion and exclusion criteria. - From the eligible
<50%group, take a uniform random sample and obtain consent until the recruitment target is reached. Exclude non-consenting or excess patients, and report HF and all-cause hospitalizations over 12 months from screening for that bin. - Install the device within 60 days of consent, and at least 14 days after discharge if the patient was recently hospitalized.
- On Day 0, the patient starts the study at the Activation date and is randomized into one of the crossover sequences.
- Sequence 1: 21 days washout (standard care), 81 days with alerts off and RPMP data off (standard care #1), 81 days with alerts off and RPMP data off (standard care #2), 21 days washout (intervention), and 162 days intervention (standard care + Heartfelt).
- Sequence 2: 21 days washout (intervention), 162 days intervention (standard care + Heartfelt), 21 days washout (standard care), 81 days with alerts off and RPMP data off (standard care #1), and 81 days with alerts off and RPMP data off (standard care #2).
- The random order can continue for up to 3.5 years using any breakdown of intervention and no-intervention periods. The example shown alternates 3-9 month intervention blocks with 3 month no-intervention blocks, then continues.
Inclusion criteria
Participants must meet the study’s full eligibility requirements. The key inclusion criteria are:
- Aged 22 years or over, male or female
- Diagnosis of chronic heart failure at least 2 months prior to randomization
- Peripheral edema exhibited on at least one HF-related hospitalization in the last 4 years
- Hospitalized for HF, received IV diuretics, or visited the ER for HF decompensation at least once in the last 6 months or twice in the last 12 months
- Treated with daily diuretics
- History of non-adherence, defined as less than 50% of data over the last 180 days, removal from an RPMP, or being considered non-adherent
- Insurance coverage that meets the IDE-B clinical trial requirements (Traditional Medicare or Medicare Advantage)
- If female and of reproductive potential, use of highly effective contraception for at least 1 month prior to screening, throughout the trial, and for 6 weeks after the end of the study
Exclusion criteria
Key exclusion criteria include:
- Bandages to the lower limbs every day
- Amputation of both feet
- Bed-bound status
- Regular wheelchair use inside the home
- Inability to take diuretics
- Participation in a conflicting study or evaluation
- Regular dialysis schedule
- History of recurrent DVT or cellulitis, defined as 2 or more episodes in the last 12 months
- Ongoing prescription of diltiazem or verapamil
- Pregnancy, or no medically approved birth control if of child-bearing potential
Study duration
- Overall study duration: 60 months
- Participant duration: 12 months randomized participation, with up to 48 months of follow-up
What are we trying to find out?
The study aims to answer the following questions:
- Is the Heartfelt device safe to use and effective at reducing HFHs?
- What is the effect of the Heartfelt device on data availability compared with existing remote monitoring devices?
- What is the effect of the Heartfelt device on HF healthcare outcomes?
Is the device safe?
Yes. The device is non-invasive and contact-free. Patients’ privacy is protected because the device only captures images of lower legs, and no personal details are stored with those images.
Who is running the study?
The study is sponsored by Heartfelt Technologies.
The Study Chair is Dr. W. H. Wilson Tang from the Cleveland Clinic.
Where is the study taking place?
Participants are recruited by Remote Patient Monitoring Companies across the United States.